Salmonella Data Preprocessing for PCSF Algorithm
By Güngör Budak
- 2 minutes read - 328 wordsThis post describes data preprocessing in Salmonella project for Prize-Collecting Steiner Forest Problem (PCSF) algorithm.
Salmonella data taken from Table S6 in Phosphoproteomic Analysis of Salmonella-Infected Cells Identifies Key Kinase Regulators and SopB-Dependent Host Phosphorylation Events by Rogers, LD et al. has been converted to tab delimited TXT file from its original XLS file for easy reading in Python.
The data should be separated into time points files (2, 5, 10 and 20 minutes) each of which will contain corresponding phophoproteins and their fold changes. Data also has another dimension which is location (membrane, nucleus, cytosol) and when we looked at each location and each time point, we saw there are not many overlapping proteins in the same time point and different locations, therefore we decided to merge location data for each time point in each protein.
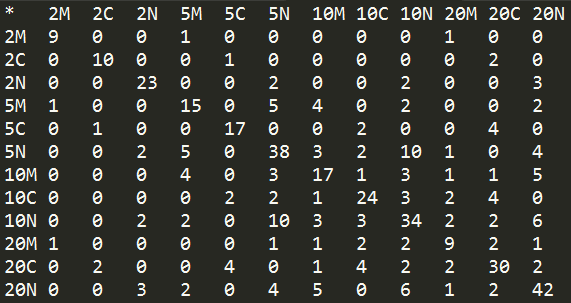
Number of overlapping proteins in each dimensions (2N; 2 min. & Nucleus; 10C: 10 min. & Cytosol).
The data table is read for each time point row by row and if the row has any data satisfying conditions (significance 0.05 and variance 15) in one or more than one locations, they are stored and the maximum fold change among them (in the case of more than one locations) is obtained and stored with HGNC name of the protein. HGNC names of the proteins are obtained by converting IPI IDs of their peptides to HGNC names of the proteins (DAVID is used for this conversion). After collecting all HGNC names and maximum fold changes, each HGNC name and corresponding maximum fold change(s) are written as an output. If there are multiple maximum fold changes for an HGNC name due to the same protein in multiple rows, then their maximum is obtained and written next to that HGNC name.
Outputs of this preprocessing are four TXT files of HGNC name – fold change pairs and each file contains proteins that are found significant and the most activated or deactivated at one time point.